Nội dung:
- 1 Quy định mới của FDA về đăng ký cơ sở thiết bị y tế năm 2026
- 2 Đối tượng bắt buộc phải đăng ký cơ sở sản xuất thiết bị y tế FDA
- 3 Các hình thức đăng ký FDA cho cơ sở thiết bị y tế
- 4 Hồ sơ đăng ký cơ sở sản xuất thiết bị y tế FDA gồm những gì
- 5 Quy trình đăng ký cơ sở sản xuất thiết bị y tế FDA 2026
- 6 Thời gian và chi phí đăng ký FDA cơ sở thiết bị y tế 2026
- 7 Dịch vụ đăng ký cơ sở sản xuất thiết bị y tế FDA 2026 tại Viện Chứng Nhận Toàn Cầu
- 8 Lợi ích khi sử dụng dịch vụ đăng ký FDA trọn gói
Đưa sản phẩm thiết bị y tế “Made in Vietnam” chinh phục thị trường Hoa Kỳ là một mục tiêu đầy tiềm năng nhưng cũng không ít thách thức. Yêu cầu bắt buộc và cũng là rào cản pháp lý lớn nhất chính là việc đăng ký cơ sở sản xuất thiết bị y tế với Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA). Tôi tin rằng nhiều doanh nghiệp cũng cảm nhận được sức nóng này, đặc biệt khi quy định mới của FDA cho năm 2026 sắp được áp dụng, khiến quy trình càng trở nên phức tạp hơn bao giờ hết. Chắc hẳn không ít đơn vị đang loay hoay không biết bắt đầu từ đâu: từ việc xác định đúng loại hình đăng ký, chuẩn bị hồ sơ, thực hiện Device Listing cho từng sản phẩm, cho đến việc tìm kiếm và chỉ định một US Agent bắt buộc. Việc hiểu sai một quy định nhỏ cũng có thể khiến toàn bộ lô hàng bị giữ lại, gây ra những tổn thất không đáng có.
Quy định mới của FDA về đăng ký cơ sở thiết bị y tế năm 2026
Bước vào năm 2026, các quy định của Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA) không chỉ là những cập nhật nhỏ lẻ mà phản ánh sự thay đổi lớn trong cách tiếp cận quản lý thiết bị y tế, tập trung vào an toàn, truy xuất nguồn gốc và hội nhập tiêu chuẩn quốc tế. Việc nắm bắt những thay đổi này không còn là một lợi thế, mà là yêu cầu bắt buộc để duy trì hoạt động kinh doanh tại thị trường Mỹ.
Cập nhật yêu cầu pháp lý FDA áp dụng cho năm 2026
Các quy định pháp lý cho năm 2026 và các năm tiếp theo sẽ xoay quanh việc siết chặt quản lý và nâng cao tiêu chuẩn. Doanh nghiệp cần đặc biệt chú ý đến các điểm sau:
- Hài hòa hóa Hệ thống Quản lý Chất lượng (QMSR): FDA đang trong giai đoạn cuối cùng của việc thay thế Quy định Hệ thống Chất lượng (QSR) hiện hành bằng Quy định Hệ thống Quản lý Chất lượng (QMSR), vốn được hài hòa hóa với tiêu chuẩn quốc tế ISO 13485:2016. Điều này có nghĩa là các nhà sản xuất sẽ phải điều chỉnh hệ thống quản lý chất lượng của mình để vừa tuân thủ yêu cầu của FDA, vừa phù hợp với tiêu chuẩn được công nhận toàn cầu.
- Tăng cường yêu cầu về An ninh mạng (Cybersecurity): Đối với các thiết bị y tế có kết nối mạng hoặc chứa phần mềm, FDA đã ban hành các hướng dẫn bắt buộc về an ninh mạng. Doanh nghiệp phải cung cấp bằng chứng về việc đã triển khai các biện pháp bảo mật mạnh mẽ để bảo vệ thiết bị khỏi các mối đe dọa mạng, đảm bảo an toàn cho bệnh nhân và tính toàn vẹn của dữ liệu.
- Thực thi đầy đủ Hệ thống Định danh Thiết bị Y tế Duy nhất (UDI): Mặc dù đã được triển khai theo từng giai đoạn, đến năm 2026, hệ thống UDI (Unique Device Identification) sẽ được áp dụng và thực thi nghiêm ngặt trên hầu hết các loại thiết bị y tế. Mỗi sản phẩm phải có một mã UDI duy nhất được dán nhãn và dữ liệu phải được nộp vào cơ sở dữ liệu GUDID của FDA.
- Phí đăng ký hàng năm (MDUFA Fee): Phí đăng ký cơ sở sản xuất sẽ tiếp tục được cập nhật hàng năm theo Đạo luật Phí sử dụng Thiết bị Y tế (MDUFA). Mức phí cho năm tài chính 2026 (bắt đầu từ 01/10/2025) sẽ được công bố vào tháng 8 năm 2025 và thường có xu hướng tăng.
Thời hạn hiệu lực và chu kỳ gia hạn đăng ký hàng năm
Đây là một trong những quy định cốt lõi nhưng cũng dễ bị bỏ sót nhất.
- Thời hạn hiệu lực: Mã số đăng ký cơ sở (FDA Establishment Registration) chỉ có hiệu lực trong một năm tài chính của chính phủ Hoa Kỳ, tức là từ ngày 01 tháng 10 năm nay đến ngày 30 tháng 9 năm sau.
- Chu kỳ gia hạn: Tất cả các cơ sở đã đăng ký bắt buộc phải thực hiện gia hạn hàng năm trong khoảng thời gian từ ngày 01 tháng 10 đến ngày 31 tháng 12. Nếu không hoàn tất việc gia hạn trong thời gian này, mã số đăng ký sẽ tự động bị vô hiệu hóa.
- Hậu quả: Khi mã số đăng ký không còn hiệu lực, mọi lô hàng thiết bị y tế của doanh nghiệp sẽ bị từ chối nhập cảnh vào Hoa Kỳ, gây ra tình trạng hàng hóa bị giữ lại tại cảng (FDA detention), phát sinh chi phí lưu kho và có thể phải tái xuất hoặc tiêu hủy.
Những điểm doanh nghiệp thường hiểu sai về quy định FDA
Sự phức tạp của quy định FDA dẫn đến nhiều hiểu lầm tai hại, có thể khiến doanh nghiệp đối mặt với rủi ro pháp lý và tài chính.
- Nhầm lẫn giữa “Đăng ký FDA” và “Phê duyệt FDA”: Đây là sai lầm phổ biến nhất. Đăng ký cơ sở (Registration) và Liệt kê thiết bị (Listing) là các thủ tục hành chính bắt buộc để FDA biết bạn là ai và bạn sản xuất/kinh doanh sản phẩm gì. Nó hoàn toàn không có nghĩa là sản phẩm của bạn đã được “FDA phê duyệt” (FDA Approved). Sự phê duyệt (ví dụ như 510(k) Clearance hoặc PMA Approval) là một quy trình đánh giá khoa học và kỹ thuật khắt khe hơn nhiều, chỉ áp dụng cho các thiết bị y tế có mức độ rủi ro từ trung bình đến cao.
- Xem nhẹ vai trò của US Agent: Nhiều doanh nghiệp cho rằng US Agent chỉ là một địa chỉ liên lạc tại Mỹ. Thực tế, US Agent là đại diện pháp lý chính thức của cơ sở nước ngoài trước FDA. Họ chịu trách nhiệm liên lạc, hỗ trợ FDA trong việc lên lịch thanh tra, và trả lời các câu hỏi liên quan đến sản phẩm. Lựa chọn một US Agent thiếu kinh nghiệm hoặc không am hiểu quy định có thể gây ra nhiều rắc rối.
- Sử dụng mã số đăng ký FDA để quảng cáo: FDA nghiêm cấm việc sử dụng logo FDA hoặc mã số đăng ký cơ sở trên nhãn sản phẩm hoặc tài liệu quảng cáo để ngụ ý rằng cơ sở hoặc sản phẩm được FDA chứng nhận hay xác nhận. Hành vi này được xem là “misbranding” (ghi nhãn sai) và có thể dẫn đến các hành động pháp lý từ FDA.

✍️ Tìm hiểu thêm: Dịch vụ Đăng ký chứng nhận FDA khẩu trang y tế
Đối tượng bắt buộc phải đăng ký cơ sở sản xuất thiết bị y tế FDA
Quy định của Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA) về đăng ký cơ sở (Establishment Registration) có phạm vi áp dụng rất rộng, không chỉ giới hạn ở nhà máy sản xuất cuối cùng. Mục tiêu của FDA là kiểm soát và truy xuất toàn bộ chuỗi cung ứng của thiết bị y tế nhập khẩu vào Mỹ, đảm bảo mọi khâu đều tuân thủ các tiêu chuẩn an toàn và chất lượng. Do đó, việc xác định đúng vai trò và trách nhiệm của doanh nghiệp là bước đầu tiên và quan trọng nhất.
Nhà sản xuất thiết bị y tế tại Việt Nam
Đây là đối tượng rõ ràng nhất và bắt buộc phải thực hiện đăng ký cơ sở sản xuất với FDA. Bất kỳ cơ sở nào tại Việt Nam tham gia vào quá trình chế tạo, sản xuất, lắp ráp, hoặc xử lý một thiết bị y tế từ nguyên liệu thô đến thành phẩm cuối cùng, nếu sản phẩm đó có mục tiêu xuất khẩu sang thị trường Hoa Kỳ, đều phải đăng ký. Điều này áp dụng cho mọi quy mô doanh nghiệp, từ các tập đoàn lớn đến các xưởng sản xuất nhỏ. Việc đăng ký không chỉ là một thủ tục hành chính mà còn là sự công nhận rằng cơ sở đó nằm trong hệ thống giám sát của FDA.
Đơn vị lắp ráp, đóng gói, dán nhãn (OEM/ODM)
Một trong những hiểu lầm phổ biến là chỉ có đơn vị sở hữu thương hiệu mới cần đăng ký. Trên thực tế, FDA yêu cầu tất cả các bên tham gia vào quá trình sản xuất phải đăng ký, bao gồm cả các đối tác OEM (Original Equipment Manufacturer) và ODM (Original Design Manufacturer). Cụ thể:
- Đơn vị lắp ráp: Ngay cả khi chỉ thực hiện công đoạn lắp ráp các linh kiện do bên khác cung cấp, cơ sở đó vẫn được coi là một nhà sản xuất và phải đăng ký.
- Đơn vị đóng gói và dán nhãn (Packer/Labeler): Các cơ sở chỉ chịu trách nhiệm đóng gói sản phẩm vào bao bì cuối cùng hoặc dán nhãn mác lên thiết bị cũng bắt buộc phải đăng ký. FDA coi việc dán nhãn (labeling) và đóng gói (packaging) là các bước sản xuất quan trọng, có thể ảnh hưởng trực tiếp đến tính an toàn, hiệu quả và thông tin của sản phẩm khi đến tay người dùng.

✍️ Tìm hiểu thêm: Thông tin Nutrition Facts nhãn thông tin dinh dưỡng đầy đủ
Doanh nghiệp xuất khẩu hoặc kinh doanh thiết bị y tế vào thị trường Hoa Kỳ
Ngoài các cơ sở sản xuất trực tiếp, một số đối tượng khác cũng có thể thuộc diện phải đăng ký, tùy thuộc vào vai trò của họ trong chuỗi cung ứng:
- Nhà phát triển thông số kỹ thuật (Specification Developer): Một công ty phát triển thiết kế và thông số kỹ thuật cho một thiết bị y tế nhưng thuê một bên khác (nhà sản xuất theo hợp đồng) để sản xuất, vẫn phải đăng ký cơ sở với FDA.
- Nhà xuất khẩu (Foreign Exporter): Nếu nhà xuất khẩu tại Việt Nam đồng thời thực hiện một trong các chức năng sản xuất như đóng gói lại, dán nhãn lại trước khi xuất hàng, họ cũng phải đăng ký với tư cách là một cơ sở sản xuất.
Các hình thức đăng ký FDA cho cơ sở thiết bị y tế
FDA phân loại các hình thức đăng ký dựa trên chức năng cụ thể của từng cơ sở trong chuỗi cung ứng. Việc lựa chọn đúng hình thức đăng ký là rất quan trọng để đảm bảo hồ sơ hợp lệ và tuân thủ đúng quy định.
Đăng ký cơ sở sản xuất (Manufacturer Registration)
Đây là hình thức đăng ký phổ biến và bao trùm nhất, áp dụng cho tất cả các cơ sở được đề cập ở trên: từ nhà sản xuất thành phẩm, nhà sản xuất linh kiện, đơn vị lắp ráp, đến các cơ sở đóng gói, dán nhãn, và thậm chí cả các cơ sở tái xử lý thiết bị y tế dùng một lần. Khi đăng ký với tư cách nhà sản xuất (manufacturer), cơ sở đó sẽ phải khai báo và niêm yết tất cả các thiết bị y tế (Device Listing) được sản xuất tại địa điểm đó.
Đăng ký cơ sở kinh doanh, phân phối (Initial Importer / Distributor)
Hình thức này áp dụng cho các đơn vị tại Hoa Kỳ. Nhà nhập khẩu ban đầu (Initial Importer) là đơn vị đầu tiên tại Mỹ đứng ra nhập khẩu thiết bị y tế từ một cơ sở nước ngoài để tiếp tục kinh doanh và phân phối tại thị trường này. Đơn vị này bắt buộc phải đăng ký cơ sở của mình với FDA. Vai trò của Initial Importer rất quan trọng trong việc truy xuất nguồn gốc sản phẩm nếu có sự cố xảy ra. Các nhà phân phối khác trong chuỗi cung ứng tại Mỹ (mua lại từ Initial Importer) thường không bắt buộc phải đăng ký, trừ khi họ thực hiện các thao tác làm thay đổi thiết bị như đóng gói lại hoặc dán nhãn lại.
Vai trò của US Agent trong hồ sơ đăng ký FDA
Đối với mọi cơ sở sản xuất nước ngoài (bao gồm cả Việt Nam), việc chỉ định một Đại diện tại Hoa Kỳ (US Agent) là yêu cầu bắt buộc trong hồ sơ đăng ký. US Agent không phải là một nhà phân phối hay đối tác thương mại, mà là một đại diện pháp lý về mặt quy định. Vai trò của US Agent bao gồm:
- Là đầu mối liên lạc chính thức giữa FDA và cơ sở sản xuất nước ngoài.
- Tiếp nhận mọi thông báo, văn bản, và thư từ từ FDA thay mặt cho doanh nghiệp.
- Hỗ trợ FDA trong việc sắp xếp các cuộc thanh tra, kiểm tra tại cơ sở sản xuất ở nước ngoài.
- Trả lời các câu hỏi của FDA liên quan đến các thiết bị y tế đã được niêm yết.
Việc lựa chọn một US Agent uy tín và có kinh nghiệm là yếu tố then chốt để đảm bảo quá trình liên lạc với FDA diễn ra suôn sẻ và tuân thủ đúng pháp luật.
Hồ sơ đăng ký cơ sở sản xuất thiết bị y tế FDA gồm những gì
Việc chuẩn bị một bộ hồ sơ đầy đủ và chính xác là nền tảng quyết định sự thành công của quá trình đăng ký cơ sở với FDA. Bất kỳ sai sót hay thiếu hụt thông tin nào cũng có thể dẫn đến sự chậm trễ, yêu cầu bổ sung, hoặc thậm chí là từ chối hồ sơ, gây ảnh hưởng trực tiếp đến kế hoạch xuất khẩu của doanh nghiệp.
Thông tin pháp lý doanh nghiệp
Đây là những thông tin cơ bản để FDA xác định danh tính và tư cách pháp nhân của đơn vị đăng ký. Hồ sơ cần thể hiện rõ ràng và nhất quán các thông tin sau:
- Tên pháp lý đầy đủ của công ty: Tên phải trùng khớp hoàn toàn với giấy phép đăng ký kinh doanh.
- Địa chỉ trụ sở chính: Địa chỉ đăng ký chính thức của doanh nghiệp, không nhất thiết phải là địa chỉ nhà xưởng.
- Thông tin người sở hữu/người điều hành (Owner/Operator): Cung cấp thông tin chi tiết về cá nhân hoặc tổ chức chịu trách nhiệm cao nhất về mặt pháp lý cho hoạt động của cơ sở.
- Thông tin người liên hệ chính thức (Official Correspondent): Người sẽ là đầu mối liên lạc chính thức với FDA, chịu trách nhiệm nhận và phản hồi mọi thông báo, yêu cầu từ cơ quan này.
Thông tin cơ sở sản xuất
Phần này tập trung vào địa điểm vật lý nơi các hoạt động liên quan đến thiết bị y tế diễn ra. FDA yêu cầu khai báo chi tiết cho từng địa điểm riêng biệt tham gia vào chuỗi sản xuất.
- Địa chỉ cụ thể của nhà máy, xưởng sản xuất: Nơi diễn ra các công đoạn chế tạo, lắp ráp, xử lý sản phẩm.
- Thông tin về các hoạt động được thực hiện tại cơ sở: Cần xác định rõ vai trò của cơ sở, ví dụ như sản xuất theo hợp đồng (Contract Manufacturer), tiệt trùng (Sterilizer), đóng gói và dán nhãn lại (Repackager/Relabeler). Mỗi vai trò có thể có những yêu cầu khai báo khác nhau.
Danh mục thiết bị y tế (Device Listing)
Bên cạnh việc đăng ký cơ sở (Establishment Registration), doanh nghiệp bắt buộc phải liệt kê tất cả các thiết bị y tế mà mình sản xuất để xuất khẩu vào Hoa Kỳ. Đây được gọi là Device Listing.
Mỗi thiết bị trong danh mục cần được khai báo với các thông tin:
- Tên thương mại (Proprietary Name): Tên gọi sản phẩm sẽ được lưu hành trên thị trường.
- Mã sản phẩm FDA (FDA Product Code): Một mã gồm 3 chữ cái và 3 chữ số do FDA quy định để phân loại thiết bị.
- Số đệ trình tiền thị trường (Premarket Submission Number): Nếu có, ví dụ như số 510(k), PMA, De Novo. Đây là yêu cầu bắt buộc đối với hầu hết các thiết bị Class II và Class III.

✍️ Tìm hiểu thêm: Dịch vụ đăng ký FDA dược phẩm và thiết bị y tế
Phân loại thiết bị y tế theo FDA
Việc xác định đúng phân loại rủi ro của thiết bị là yêu cầu tiên quyết trước khi tiến hành đăng ký, vì nó ảnh hưởng trực tiếp đến các yêu cầu pháp lý đi kèm. FDA chia thiết bị y tế thành 3 nhóm chính:
- Class I (Rủi ro thấp): Các thiết bị có rủi ro tối thiểu đối với người dùng, ví dụ như băng gạc y tế, găng tay khám bệnh không vô trùng, bàn chải đánh răng. Hầu hết các thiết bị này được miễn trừ yêu cầu đệ trình tiền thị trường.
- Class II (Rủi ro trung bình): Nhóm phổ biến nhất, bao gồm các thiết bị như kim tiêm, máy đo huyết áp, kính áp tròng. Đa số thiết bị Class II yêu cầu phải có giấy phép 510(k) trước khi được lưu hành.
- Class III (Rủi ro cao): Các thiết bị hỗ trợ hoặc duy trì sự sống, cấy ghép vào cơ thể, có nguy cơ gây thương tổn nghiêm trọng. Ví dụ: máy tạo nhịp tim, van tim nhân tạo. Các thiết bị này yêu cầu quy trình phê duyệt nghiêm ngặt nhất là Premarket Approval (PMA).
Thông tin US Agent bắt buộc
Đối với tất cả các cơ sở sản xuất nằm ngoài lãnh thổ Hoa Kỳ, việc chỉ định một Đại diện tại Hoa Kỳ (US Agent) là yêu cầu bắt buộc.
- Vai trò của US Agent: Là cầu nối liên lạc chính thức giữa FDA và doanh nghiệp nước ngoài. Họ chịu trách nhiệm trả lời các câu hỏi từ FDA, hỗ trợ sắp xếp các cuộc thanh tra, và tiếp nhận các thông tin, tài liệu pháp lý.
- Yêu cầu đối với US Agent: Phải là một cá nhân, công ty hoặc tổ chức có địa chỉ cư trú hoặc kinh doanh thực tế tại Hoa Kỳ (không chấp nhận địa chỉ hộp thư P.O. Box). Thông tin liên lạc của US Agent phải được cung cấp đầy đủ và chính xác trong hồ sơ đăng ký.
Quy trình đăng ký cơ sở sản xuất thiết bị y tế FDA 2026
Quy trình đăng ký cơ sở sản xuất thiết bị y tế với FDA là một chuỗi các bước được chuẩn hóa, đòi hỏi sự chính xác tuyệt đối trong từng giai đoạn. Việc tuân thủ đúng trình tự không chỉ giúp doanh nghiệp hoàn thành nghĩa vụ pháp lý mà còn đảm bảo quá trình xuất khẩu diễn ra suôn sẻ, tránh các rủi ro bị từ chối hoặc tạm giữ hàng hóa tại cảng Hoa Kỳ.
Bước 1: Rà soát loại hình cơ sở và thiết bị y tế
Đây là bước nền tảng và có ý nghĩa quyết định đến toàn bộ quy trình. Sai sót ở giai đoạn này có thể dẫn đến việc chuẩn bị sai hồ sơ và lãng phí thời gian, chi phí. Doanh nghiệp cần xác định rõ hai yếu tố chính:
- Loại hình cơ sở (Establishment Type): Doanh nghiệp của bạn hoạt động với vai trò nào? Là nhà sản xuất (Manufacturer), đơn vị chỉ đóng gói và dán nhãn lại (Repackager/Relabeler), hay nhà sản xuất theo hợp đồng (Contract Manufacturer)? Mỗi loại hình sẽ có những yêu cầu khai báo và trách nhiệm pháp lý khác nhau. Việc xác định sai có thể dẫn đến vi phạm quy định của FDA.
- Phân loại thiết bị y tế (Device Classification): Thiết bị của bạn thuộc Phân loại I, II, hay III? Mức độ rủi ro của thiết bị sẽ quyết định lộ trình pháp lý cần tuân thủ. Ví dụ, hầu hết thiết bị Loại I (Class I) được miễn trừ Thông báo trước khi đưa ra thị trường (Premarket Notification 510(k)), trong khi phần lớn thiết bị Loại II (Class II) bắt buộc phải có 510(k). Thiết bị Loại III (Class III), với mức độ rủi ro cao nhất, thường yêu cầu quy trình Phê duyệt trước khi đưa ra thị trường (Premarket Approval – PMA) phức tạp hơn nhiều.
Việc rà soát chính xác mã sản phẩm (product code) và các quy định áp dụng cho từng loại thiết bị là yêu cầu bắt buộc để định hướng đúng các bước tiếp theo.
Bước 2: Chuẩn bị hồ sơ theo đúng yêu cầu FDA
Sau khi đã xác định rõ loại hình và phân loại, doanh nghiệp cần tập hợp một bộ hồ sơ đầy đủ và nhất quán. Các thông tin cung cấp cho FDA phải chính xác tuyệt đối và đồng bộ trên mọi tài liệu.
Hồ sơ đăng ký cơ sở thường bao gồm:
- Thông tin pháp lý doanh nghiệp: Tên công ty, địa chỉ đăng ký kinh doanh, thông tin liên hệ của người chịu trách nhiệm. Đặc biệt, doanh nghiệp phải có số DUNS (Data Universal Numbering System) – một mã số định danh doanh nghiệp toàn cầu.
- Thông tin cơ sở sản xuất: Địa chỉ vật lý của nhà máy, xưởng sản xuất nơi thiết bị được tạo ra.
- Thông tin Đại diện tại Hoa Kỳ (US Agent): Đối với các cơ sở ngoài Hoa Kỳ, việc chỉ định một US Agent là bắt buộc. US Agent đóng vai trò là cầu nối liên lạc chính thức giữa doanh nghiệp và FDA.
- Danh mục thiết bị y tế (Device Listing): Liệt kê toàn bộ các thiết bị y tế mà cơ sở sản xuất và dự định xuất khẩu vào thị trường Hoa Kỳ. Mỗi thiết bị phải được khai báo với mã sản phẩm tương ứng.
Tất cả thông tin phải được trình bày bằng tiếng Anh. Bất kỳ sự thiếu sót hay không nhất quán nào cũng có thể là nguyên nhân khiến hồ sơ bị trì hoãn hoặc từ chối.
Bước 3: Khai báo và nộp hồ sơ trên hệ thống FDA
Toàn bộ quy trình đăng ký và niêm yết thiết bị được thực hiện trực tuyến thông qua hệ thống của FDA.
Đầu tiên, doanh nghiệp cần tạo tài khoản trên hệ thống FDA Unified Registration and Listing System (FURLS). Sau khi có tài khoản, bước tiếp theo là thanh toán phí đăng ký thường niên (Annual Registration Fee). Mức phí này được FDA cập nhật hàng năm và việc thanh toán là điều kiện tiên quyết để có thể nộp hồ sơ.
Khi đã hoàn tất thanh toán, doanh nghiệp sẽ tiến hành khai báo trực tuyến các thông tin đã chuẩn bị ở Bước 2. Hệ thống FURLS có giao diện khá phức tạp và đòi hỏi người khai báo phải hiểu rõ các thuật ngữ chuyên ngành cũng như yêu cầu của FDA. Việc nhập sai thông tin có thể dẫn đến việc đăng ký không hợp lệ.
Bước 4: Nhận mã số đăng ký FDA và xác nhận Device Listing
Sau khi FDA xem xét và chấp thuận hồ sơ, hệ thống sẽ tự động cấp các mã số quan trọng:
- Mã số đăng ký cơ sở (Establishment Registration Number): Xác nhận cơ sở của bạn đã được đăng ký trong hệ thống của FDA.
- Mã số niêm yết thiết bị (Device Listing Number): Cấp riêng cho từng loại thiết bị y tế đã được khai báo.
Doanh nghiệp cần lưu ý rằng, việc nhận được các mã số này không đồng nghĩa với “chứng nhận FDA” hay “phê duyệt sản phẩm. Đây chỉ là sự xác nhận rằng cơ sở đã hoàn thành nghĩa vụ đăng ký với cơ quan quản lý. Sau khi có các mã số này, doanh nghiệp có thể bắt đầu các hoạt động xuất khẩu hợp pháp vào Hoa Kỳ. Đồng thời, doanh nghiệp phải thực hiện gia hạn hàng năm trong khoảng thời gian từ ngày 1 tháng 10 đến ngày 31 tháng 12 để duy trì hiệu lực đăng ký.
Thời gian và chi phí đăng ký FDA cơ sở thiết bị y tế 2026
Việc lập kế hoạch chi tiết về thời gian và ngân sách là yếu tố then chốt quyết định sự thành công khi đưa thiết bị y tế vào thị trường Hoa Kỳ. Hiểu rõ các khoản phí bắt buộc và các chi phí phát sinh sẽ giúp doanh nghiệp chủ động trong việc chuẩn bị nguồn lực và tránh những gián đoạn không đáng có.
Thời gian xử lý hồ sơ đăng ký FDA
Nhiều doanh nghiệp thường lầm tưởng rằng thời gian đăng ký FDA kéo dài hàng tháng trời. Trên thực tế, thời gian xử lý của FDA sau khi nhận đủ hồ sơ và thanh toán là tương đối nhanh, thường chỉ trong vòng vài ngày làm việc đến một tuần. Hệ thống điện tử của FDA (FURLS – FDA Unified Registration and Listing System) cho phép quá trình này diễn ra hiệu quả.
Tuy nhiên, khoảng thời gian quan trọng nhất và thường kéo dài nhất chính là giai đoạn chuẩn bị. Giai đoạn này bao gồm các công việc như:
- Xác định chính xác loại hình cơ sở (nhà sản xuất, đóng gói, nhà xuất khẩu…).
- Phân loại đúng mã sản phẩm cho từng thiết bị y tế (Device Listing).
- Tìm kiếm và ký hợp đồng với một US Agent (Đại diện tại Hoa Kỳ) uy tín.
- Tập hợp và chuẩn hóa toàn bộ thông tin pháp lý của doanh nghiệp.
Quá trình chuẩn bị này có thể mất từ vài tuần đến vài tháng, tùy thuộc vào mức độ phức tạp của sản phẩm và sự sẵn có của hồ sơ nội bộ. Do đó, doanh nghiệp cần bắt đầu quy trình từ sớm để đảm bảo kịp thời cho các kế hoạch xuất khẩu.
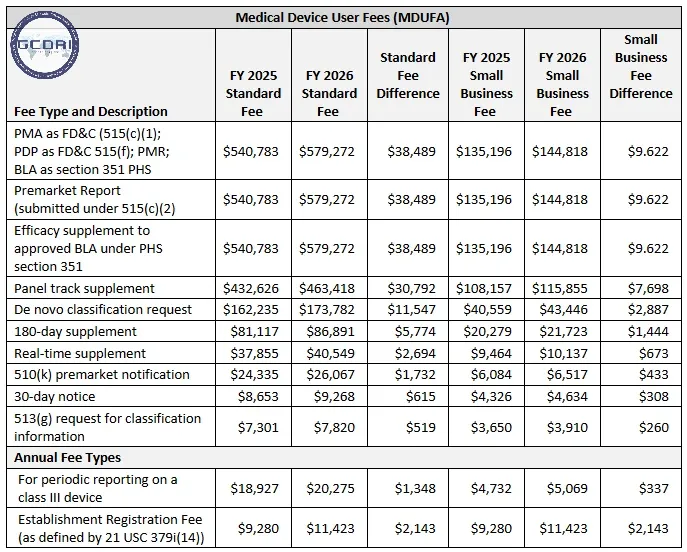
Lập kế hoạch ngân sách cho các khoản phí đăng ký FDA là bước quan trọng
Phí đăng ký FDA theo quy định hiện hành
Khoản phí quan trọng nhất là Phí Đăng ký Cơ sở Thường niên (Annual Establishment Registration Fee). Đây là khoản phí bắt buộc mà mọi cơ sở sản xuất, kinh doanh thiết bị y tế phải nộp cho FDA mỗi năm để duy trì hiệu lực đăng ký.
- Mức phí thay đổi hàng năm: FDA công bố mức phí mới cho mỗi năm tài chính (bắt đầu từ ngày 1 tháng 10) vào khoảng tháng 8 hàng năm. Ví dụ, phí cho năm tài chính 2024 là $6,493. Doanh nghiệp cần cập nhật thông tin trên trang web chính thức của FDA để biết mức phí chính xác cho năm 2026.
- Thời hạn nộp phí: Phí phải được thanh toán trong khoảng thời gian từ ngày 1 tháng 10 đến ngày 31 tháng 12 hàng năm để gia hạn đăng ký cho năm tiếp theo. Đối với đăng ký lần đầu, phí phải được nộp trước khi FDA xử lý hồ sơ.
- Phí không hoàn lại: Khoản phí này sẽ không được hoàn lại trong bất kỳ trường hợp nào, kể cả khi doanh nghiệp ngừng hoạt động hoặc hồ sơ bị từ chối do sai sót.
Các chi phí phát sinh doanh nghiệp cần lưu ý
Ngoài phí đăng ký chính thức nộp cho FDA, doanh nghiệp cần dự trù ngân sách cho các chi phí liên quan khác, vốn chiếm một phần không nhỏ trong tổng chi phí.
- Phí dịch vụ US Agent: Đây là một chi phí bắt buộc và được duy trì hàng năm. Vai trò của US Agent là làm cầu nối liên lạc giữa doanh nghiệp và FDA. Mức phí này thay đổi tùy thuộc vào nhà cung cấp dịch vụ, dao động từ vài trăm đến vài nghìn đô la mỗi năm.
- Phí tư vấn và chuẩn bị hồ sơ: Để đảm bảo hồ sơ chính xác, tuân thủ đầy đủ các quy định phức tạp và tránh rủi ro bị từ chối, nhiều doanh nghiệp lựa chọn sử dụng dịch vụ tư vấn chuyên nghiệp. Chi phí này bao gồm việc rà soát, hướng dẫn chuẩn bị giấy tờ, thực hiện khai báo trên hệ thống FDA.
- Chi phí cho các yêu cầu theo phân loại thiết bị: Việc đăng ký cơ sở và niêm yết thiết bị (Device Listing) chỉ là bước đầu tiên. Đối với các thiết bị y tế thuộc Phân loại II (Class II) và Phân loại III (Class III), doanh nghiệp sẽ phải tốn thêm chi phí đáng kể cho các quy trình phức tạp hơn như Thông báo trước khi đưa ra thị trường 510(k) hoặc Phê duyệt trước khi đưa ra thị trường (PMA). Các chi phí này cao hơn rất nhiều so với phí đăng ký cơ sở.
- Chi phí nội bộ: Bao gồm chi phí nhân sự phụ trách dự án, chi phí dịch thuật công chứng tài liệu, và các chi phí hành chính khác để phục vụ cho quá trình đăng ký.
Dịch vụ đăng ký cơ sở sản xuất thiết bị y tế FDA 2026 tại Viện Chứng Nhận Toàn Cầu
Quá trình đăng ký FDA cho cơ sở sản xuất thiết bị y tế không chỉ là một thủ tục hành chính mà còn là một cam kết pháp lý phức tạp, đòi hỏi sự am hiểu sâu sắc về các quy định của Hoa Kỳ. Đối với nhiều doanh nghiệp Việt Nam, việc tự mình điều hướng qua “mê cung” các yêu cầu này có thể dẫn đến sai sót, tốn kém thời gian và thậm chí là thất bại. Do đó, việc tìm đến một đơn vị tư vấn chuyên nghiệp như Viện Chứng Nhận Toàn Cầu là một giải pháp chiến lược, giúp đảm bảo quá trình diễn ra suôn sẻ và tuân thủ tuyệt đối.
Vai trò tư vấn chuyên môn và trách nhiệm pháp lý
Một đơn vị tư vấn chuyên nghiệp không chỉ đơn thuần là người điền hộ hồ sơ. Vai trò của họ sâu rộng hơn nhiều, bắt đầu từ việc phân tích và đánh giá toàn diện mô hình hoạt động của doanh nghiệp. Dựa trên đó, họ sẽ đưa ra chiến lược tuân thủ phù hợp nhất, từ việc xác định chính xác loại hình cơ sở cần đăng ký (nhà sản xuất, nhà đóng gói, hay nhà nhập khẩu ban đầu) đến việc phân loại thiết bị y tế theo hệ thống của FDA.
Sự tư vấn này giúp doanh nghiệp tránh được những sai lầm phổ biến, chẳng hạn như đăng ký sai loại hình hoặc khai báo thiếu thông tin, những lỗi có thể dẫn đến việc hồ sơ bị từ chối hoặc hàng hóa bị giữ lại tại cảng. Hơn nữa, đơn vị tư vấn còn đóng vai trò là cầu nối pháp lý, đặc biệt trong việc chỉ định và quản lý US Agent – đại diện pháp lý bắt buộc của doanh nghiệp tại Hoa Kỳ, chịu trách nhiệm liên lạc và xử lý các vấn đề phát sinh với FDA.
Hỗ trợ trọn gói từ tư vấn đến hoàn tất hồ sơ
Dịch vụ đăng ký FDA trọn gói được thiết kế để giảm thiểu gánh nặng cho doanh nghiệp, cho phép họ tập trung vào hoạt động sản xuất kinh doanh cốt lõi. Quy trình hỗ trợ này bao gồm tất cả các khâu cần thiết:
- Rà soát ban đầu: Đánh giá tính đầy đủ và hợp lệ của các tài liệu pháp lý, thông tin sản phẩm của doanh nghiệp.
- Chuẩn bị hồ sơ: Hướng dẫn và hỗ trợ doanh nghiệp hoàn thiện mọi giấy tờ cần thiết theo tiêu chuẩn FDA, bao gồm thông tin cơ sở, danh mục sản phẩm (Device Listing), và thông tin US Agent.
- Nộp hồ sơ và thanh toán: Thay mặt doanh nghiệp thực hiện các thao tác khai báo trên hệ thống điện tử của FDA, đồng thời xử lý việc thanh toán phí đăng ký hàng năm theo đúng quy định.
- Theo dõi và nhận kết quả: Liên tục theo dõi tiến trình xử lý hồ sơ, trao đổi với FDA khi có yêu cầu bổ sung thông tin và bàn giao mã số đăng ký FDA chính thức cho doanh nghiệp sau khi được phê duyệt.
Cam kết tuân thủ đúng quy định FDA mới nhất
Quy định của FDA không ngừng được cập nhật và thay đổi. Việc tuân thủ các quy định mới nhất, đặc biệt là các yêu cầu cho năm 2026, là yếu tố sống còn để duy trì quyền tiếp cận thị trường Hoa Kỳ. Một đơn vị tư vấn uy tín sẽ luôn cập nhật những thay đổi này và áp dụng chúng vào quá trình chuẩn bị hồ sơ. Điều này đảm bảo rằng hồ sơ của doanh nghiệp không chỉ hợp lệ tại thời điểm nộp mà còn duy trì tính tuân thủ trong dài hạn, giúp việc gia hạn đăng ký hàng năm trở nên đơn giản và nhanh chóng.
Lợi ích khi sử dụng dịch vụ đăng ký FDA trọn gói
Tiết kiệm thời gian và chi phí
Thoạt nhìn, việc tự thực hiện đăng ký có vẻ tiết kiệm chi phí. Tuy nhiên, thực tế lại thường ngược lại. Thời gian mà đội ngũ nhân sự phải bỏ ra để nghiên cứu, tìm hiểu quy định, chuẩn bị hồ sơ và xử lý các vấn đề phát sinh thường rất lớn. Bất kỳ sai sót nào cũng có thể dẫn đến việc hồ sơ bị trả lại, làm trì hoãn kế hoạch xuất khẩu và phát sinh chi phí không đáng có. Dịch vụ trọn gói giúp tối ưu hóa quy trình, thực hiện đúng ngay từ đầu, qua đó tiết kiệm thời gian và ngăn ngừa các chi phí ẩn liên quan đến sai sót và chậm trễ.
Hạn chế rủi ro pháp lý
Đây là lợi ích quan trọng nhất. Một hồ sơ đăng ký không chính xác hoặc không đầy đủ có thể gây ra những hậu quả nghiêm trọng. Hàng hóa có thể bị Cục Hải quan và Biên phòng Hoa Kỳ (CBP) từ chối nhập khẩu, dẫn đến tổn thất tài chính nặng nề. Trong trường hợp nghiêm trọng hơn, doanh nghiệp có thể bị đưa vào danh sách cảnh báo của FDA. Việc hợp tác với một đơn vị chuyên nghiệp giúp hạn chế rủi ro pháp lý ở mức tối đa, đảm bảo mọi hoạt động của doanh nghiệp đều nằm trong khuôn khổ pháp luật Hoa Kỳ.
Hồ sơ chuẩn, dễ gia hạn các năm tiếp theo
Một bộ hồ sơ được xây dựng bài bản, chính xác và đầy đủ ngay từ lần đầu tiên sẽ tạo ra một nền tảng vững chắc cho tương lai. Thông tin được khai báo nhất quán và chuẩn hóa giúp quá trình gia hạn đăng ký FDA hàng năm (diễn ra từ ngày 1 tháng 10 đến ngày 31 tháng 12) trở nên thuận lợi hơn rất nhiều. Doanh nghiệp sẽ không phải đối mặt với những yêu cầu xác minh thông tin phức tạp hoặc các rắc rối không cần thiết, đảm bảo hoạt động kinh doanh tại thị trường Mỹ không bị gián đoạn.
VIỆN NGHIÊN CỨU PHÁT TRIỂN CHỨNG NHẬN TOÀN CẦU
Địa chỉ VPGD: BT164 Central St, Khu Sunrise L, KDT The Manor Central Park, Phường Định Công, TP. Hà Nội.
Hotline: 0904.889.859
Email: lienhe@chungnhantoancau.vn
Website: https://chungnhantoancau.vn/
- Nguyễn Tiệp

- Nguyễn Tiệp

- Nguyễn Tiệp

- Nguyễn Tiệp
Like fanpage GCDRI để nhận tin mới mỗi ngày!